NF1とは

NF1とは?
カフェ・オ・レ斑と神経線維腫を主徴とし、骨、神経系、眼、皮膚等に多様な症状がみられる常染色体顕性の遺伝性疾患です1)。
- 神経線維腫症2型(NF2)は、原因遺伝子も症状も異なる全く別の疾患です。
- 難病情報センター:神経線維腫症Ⅰ型(指定難病34), https://www.nanbyou.or.jp/entry/3991, 2024/08/01確認
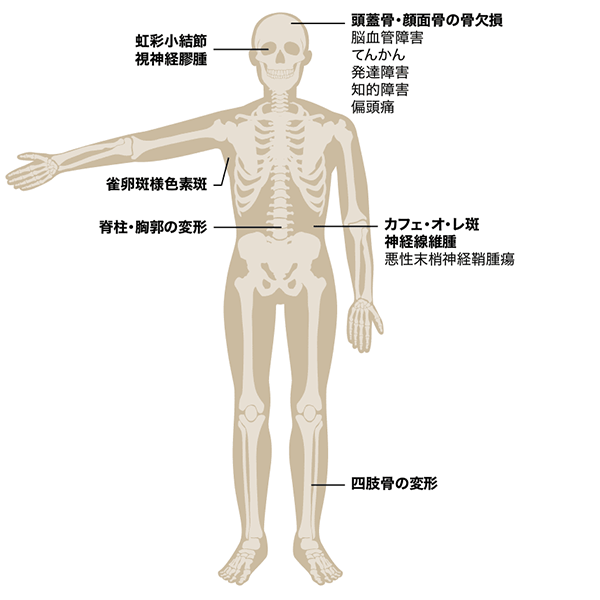
図 NF1の主な症状
原因2)
17番染色体長腕(17q11.2)にあるニューロフィブロミンをコードするNF1遺伝子の病的バリアントが原因と考えられています3)。
NF1遺伝子は神経堤※由来の細胞・器官(骨やメラノサイト、神経等)の分化に関わっています4)5)。また、細胞増殖及び細胞死抑制を促進するRASの機能を抑制しています。
NF1の患者さんではNF1遺伝子の機能が阻害され、神経堤由来の細胞・器官の分化に異常が起こること、RASの活性化により、細胞増殖と細胞死抑制が起こることからさまざまな病変が生じると推測されています6)。
- 脊椎動物の発生初期に神経管の背側から出現する一過性の移動性多能性細胞集団であり、末梢神経系、副腎髄質、メラノサイト、骨格細胞、結合細胞等多種多様な種類の細胞を生み出す7)。
図 神経堤由来の細胞・器官の例
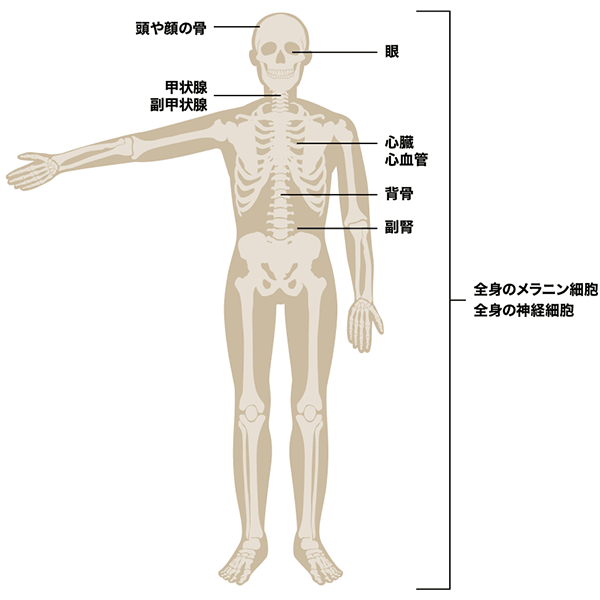
図 RASの活性化と細胞増殖(模式図)
NF1遺伝子の機能が阻害されると、遺伝子産物であるニューロフィブロミンの機能が阻害され、RASの活性化により細胞増殖と細胞死抑制が生じます。
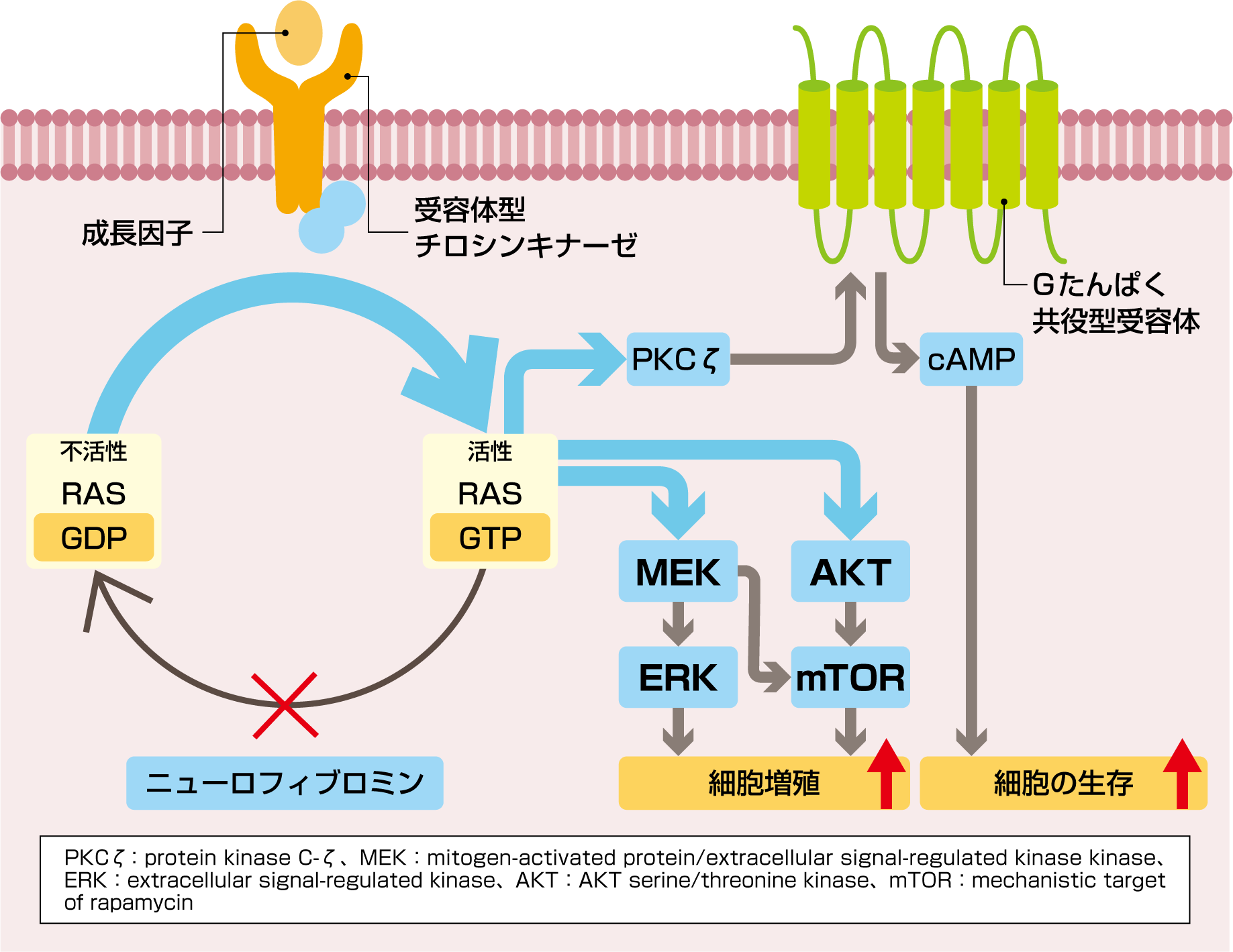
Translated by permission from Springer Nature: Nat Rev Dis Primers (Neurofibromatosis type 1, Gutmann DH et al.), COPYRIGHT(2017)
- 倉持 朗: 小児科 58(10): 1177-1194, 2017
- Gutmann DH. et al.: Proc Natl Acad Sci USA 88(21): 9658-9662, 1991
- Gitler AD. et al.: Nat Genet 33(1): 75-79, 2003
- Mazuelas H. et al.: Stem Cell Res 49: 102068, 2020
- Shyamala K. et al.: J Oral Maxillofac Pathol 19(2): 221-229, 2015
- Takahashi Y. et al.: Science 341(6148): 860-863, 2013
疫学
本邦では、出生約2,500~3,500人に1人の割合で発症8)9)し、性別や人種による差はありません1)。
本邦での推定患者数は約40,000人です10)。
- 難病情報センター: 神経線維腫症Ⅰ型(指定難病34), https://www.nanbyou.or.jp/entry/3991, 2024/08/01確認
- Anderson JL. et al.: Handb Clin Neurol 132: 75-86, 2015
- Rasmussen SA. et al.: Am J Hum Genet 68(5): 1110-1118, 2001
- 高木 廣文ほか: 厚生省特定疾患神経皮膚症候群調査研究 昭和62年度研究報告書: 11-15, 1988
診断11)
通常、臨床症状により診断を行います。
以下の臨床的診断基準のうち、2項目以上あてはまれば、NF1と診断します。詳細は、「NF1の診断と重症度分類」をご参照ください。
- 6個以上のカフェ・オ・レ斑
- 2個以上の神経線維腫(皮膚の神経線維腫や神経の神経線維腫等)またはびまん性神経線維腫
- 腋窩あるいは鼠径部の雀卵斑様色素斑(freckling)
- 視神経膠腫(optic glioma)
- 2個以上の虹彩小結節(Lisch nodule)
- 特徴的な骨病変の存在(脊柱・胸郭の変形、四肢骨の変形、頭蓋骨・顔面骨の骨欠損)
- 家系内(第一度近親者)に同症
- 神経線維腫症1型診療ガイドライン改定委員会(編). 日皮会誌 128(1): 17-34, 2018
治療
2024年7月時点では、NF1の根治療法はありません。そのため、症状に応じた診療科へ紹介し、必要に応じて各種対症療法が行われます11)。症状のひとつである叢状神経線維腫に対しては、経口治療薬であるセルメチニブ(コセルゴ®)が使用できる場合があります。詳細は電子添文をご確認ください。
- 神経線維腫症1型診療ガイドライン改定委員会(編). 日皮会誌 128(1): 17-34, 2018
定期的な経過観察の重要性11)
症状によって発現時期が異なり、発現する症状や重症度の個人差も大きく、病変によっては悪性化のリスクもあるため、定期的に診察を行う必要があります。
小児期は半年~1年に1回程度、成人は1年~数年に1回程度が目安になります。
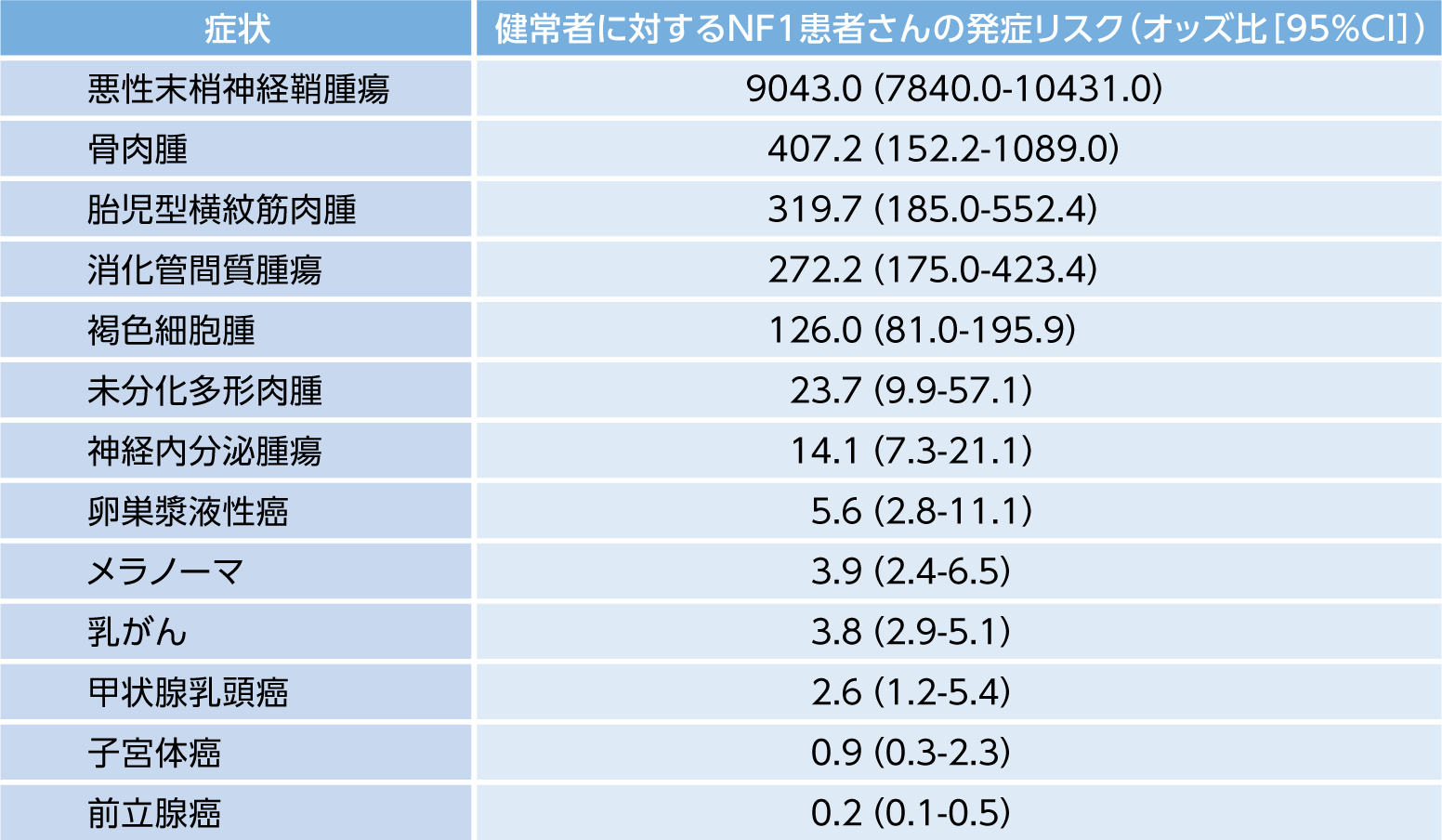
また、NF1患者では、悪性腫瘍の発症リスクが健常人と比較して2.7倍高いとされます(海外データ)12)。
各悪性腫瘍の健常者と比較した発症リスクは下記のとおりです(海外データ)13)。
- 神経線維腫症1型診療ガイドライン改定委員会(編). 日皮会誌 128(1): 17-34, 2018
- Walker L. et al.: Br J Cancer 95(2): 233-238, 2006
- Landry JP. et al.: JAMA Network Open 4(3): e210945, 2021